News
News · Proton–Electron Cosmology — cosmic-web sim, article & PDF
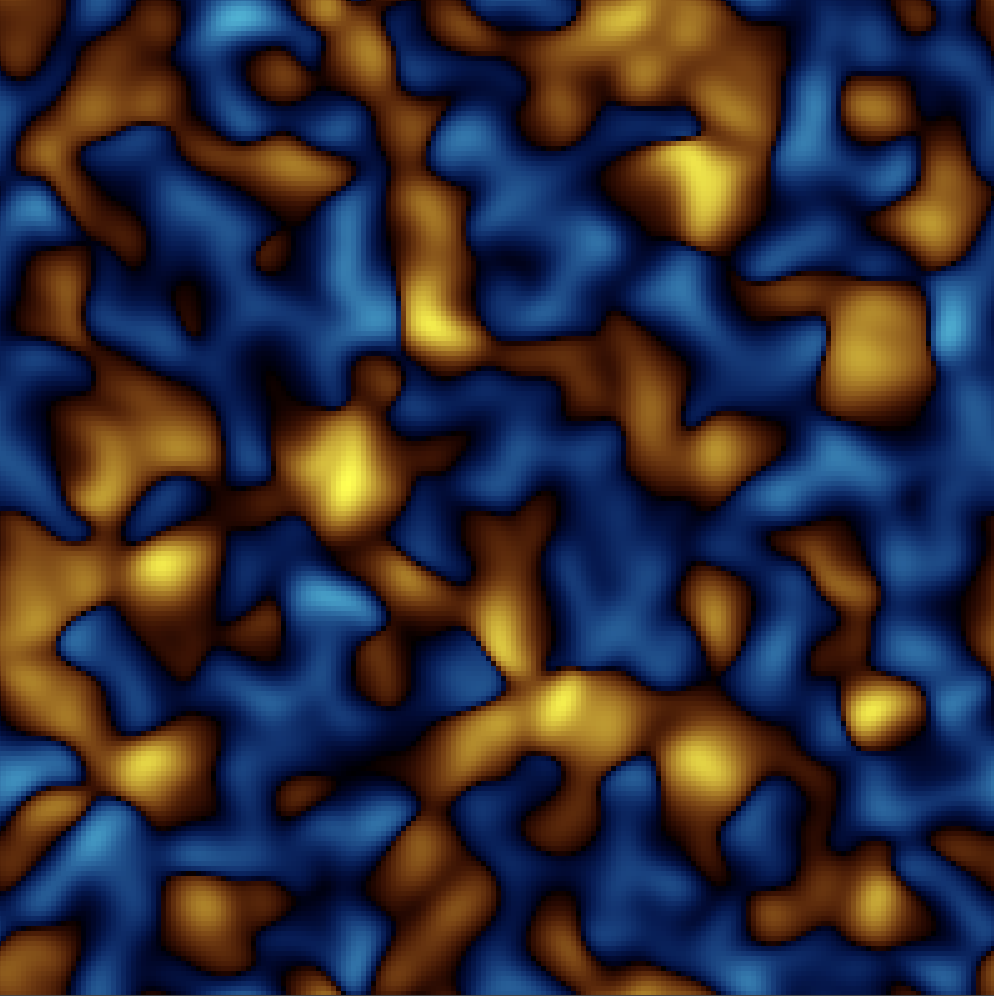
News · Charge from an electric-potential fluctuation (2D) — the contrast to the mass web
News · Foundations note: Where E = mc² comes from
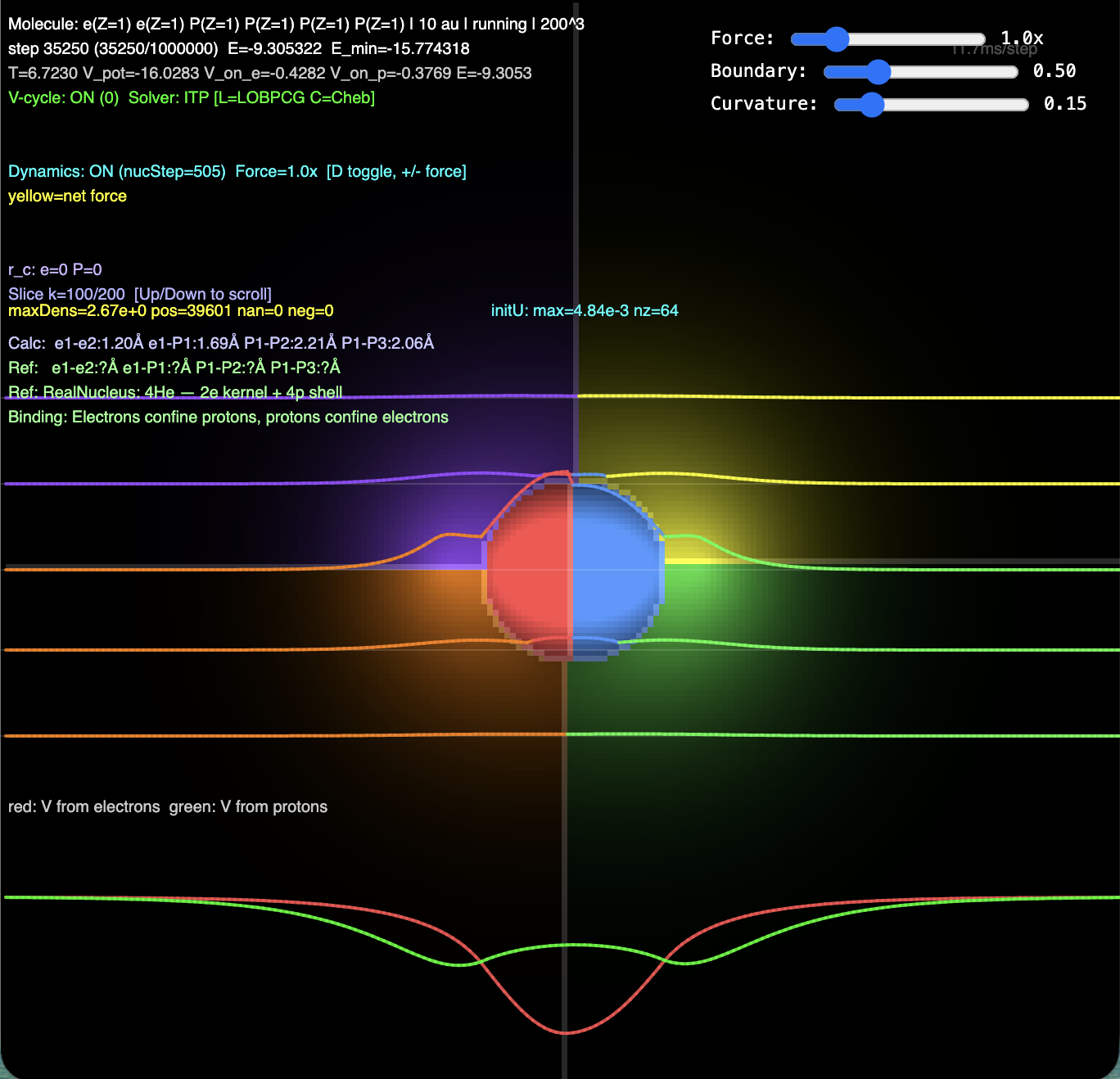
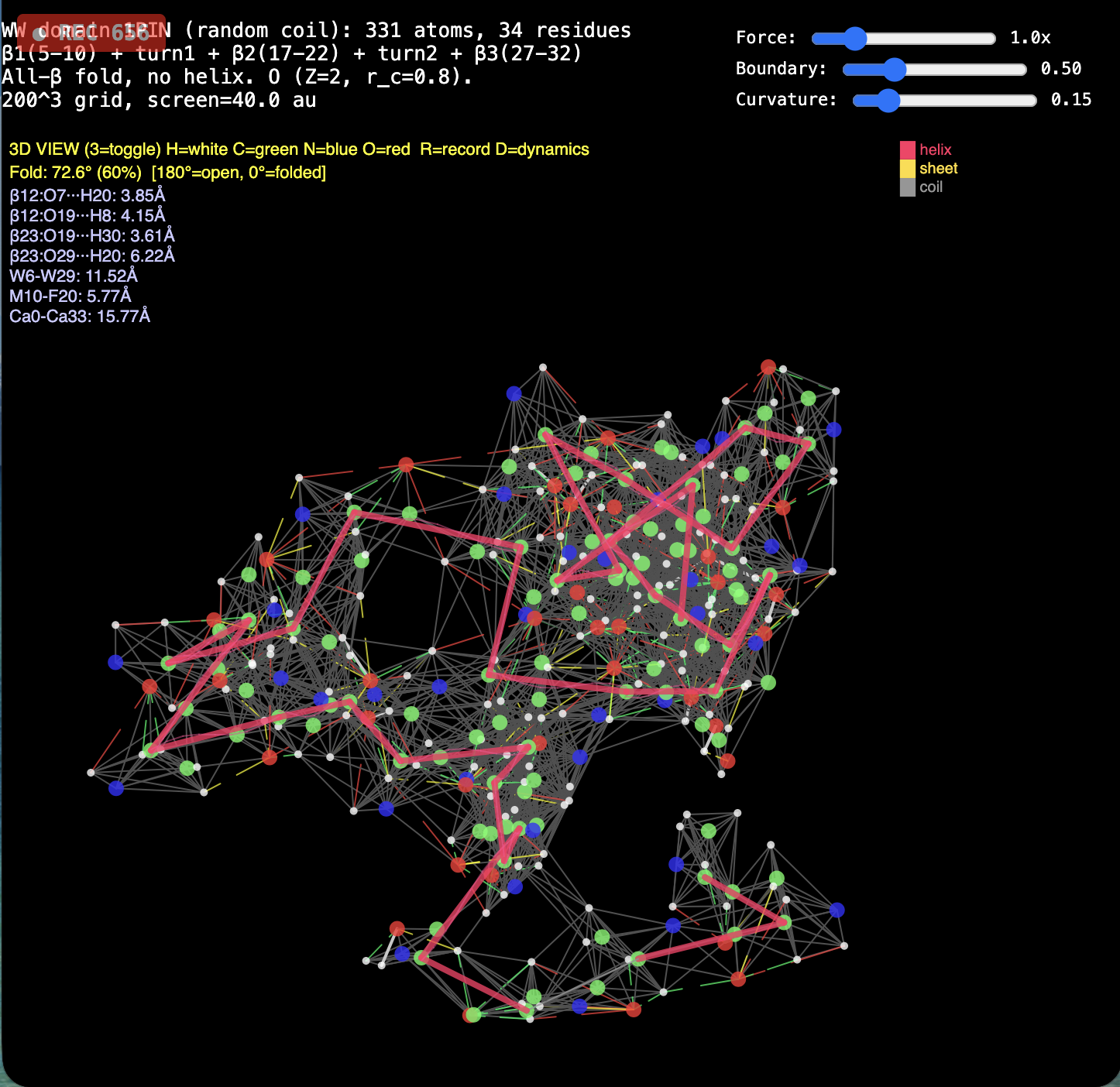
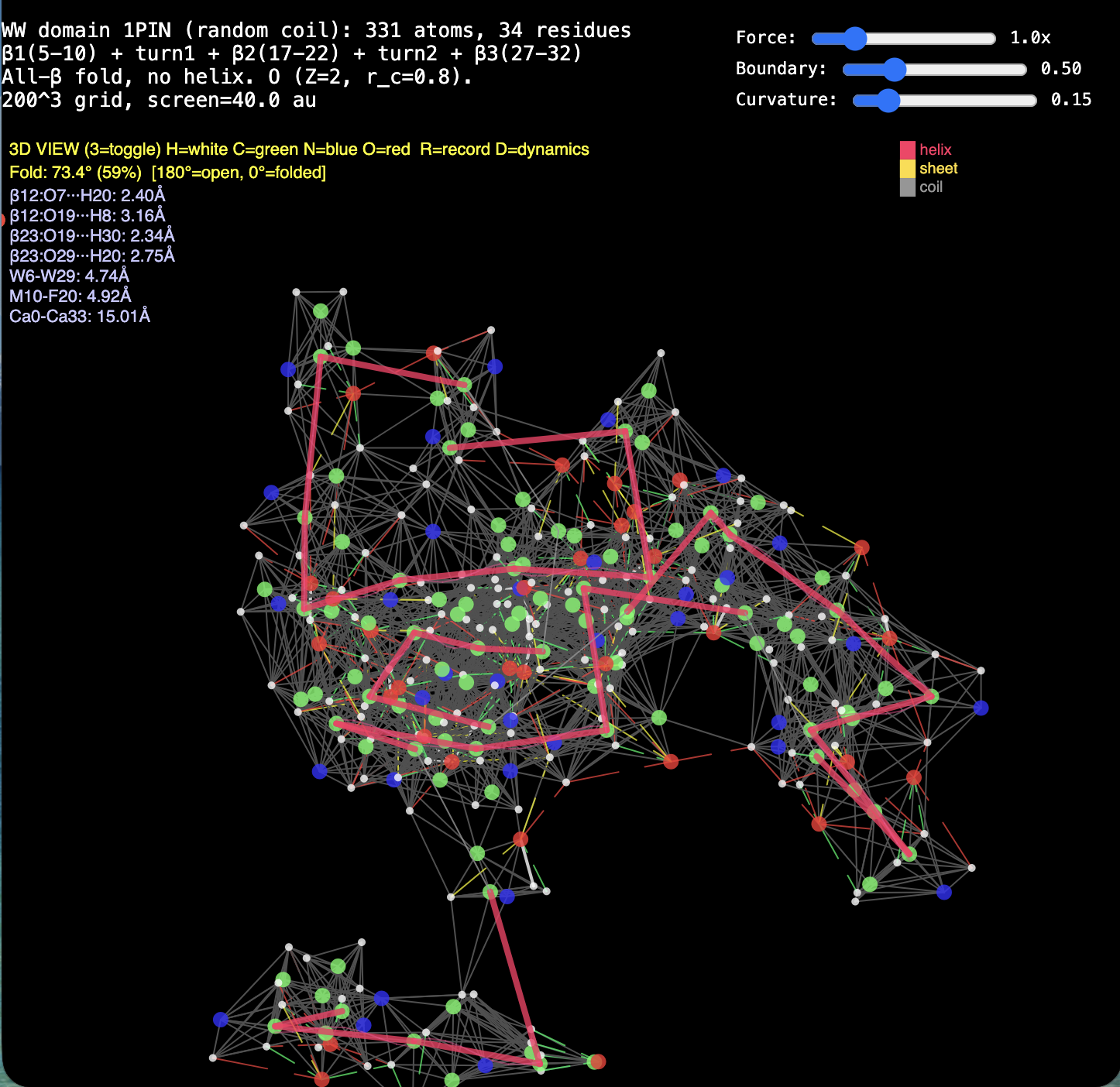
News · RealQM vs DFT for Protein Folding
Validation · No parameters to hide behind — agreement confirms the model, failure is diagnostic
Agreement ⇒ correct in that setting. No model is complete; but a model that fits many diverse observations across a setting is a correct effective description of that setting — it cannot be wrong there (as Newtonian mechanics is not wrong in its macroscopic, low-speed domain, only incomplete outside it). RealQM's untuned agreement across the periodic table, nuclear binding ratios, hydrogen bonds and reactive addition therefore makes it a correct effective model in those settings.
Every model has domain boundaries. Being incomplete, its settings have edges. A failure inside a validated setting is almost surely numerical or setup (grid, wrong minimum, geometry) — because the foundation is confirmed there; a failure in an untested setting (e.g. dispersion) simply marks a domain edge, not a contradiction.
Correct, not unique. Agreement establishes RealQM as a valid, parsimonious effective account in those settings — not the sole truth: standard QM fits the same data, so empirical agreement under-determines the mechanism. The honest claim is “RealQM is correct where it fits many,” not “QM is wrong.”
The upshot: because there are no parameters to hide behind, agreement confirms the model in-domain and failure is diagnostic — a sharpness a fitted method (which can always be re-tuned) cannot offer.
Validation · Measured, not computed — the benchmark owes nothing to any model
News · What validation means — two concrete cases (periodic table, protein folding)
1. The periodic table. RealQM: one parameter-free ab-initio code, no per-element tuning, no fitted functional, computes the whole PT in minutes to ~1–2% — the discriminating win is ionization-energy periodicity (right trend/ordering across dozens of elements from one scheme). StdQM: the ~0.1% “table values” are a per-element tuned patchwork (relativistic, multireference, ECPs…) mixed with experimental values — precision of a catalogue, not of a single prediction. Verdict: for “can a fixed theory predict the periodic table,” RealQM is the only entrant; StdQM has no single-method counterpart. (Coarse, though: 1–2% shows the model is structurally right, not chemically accurate.)
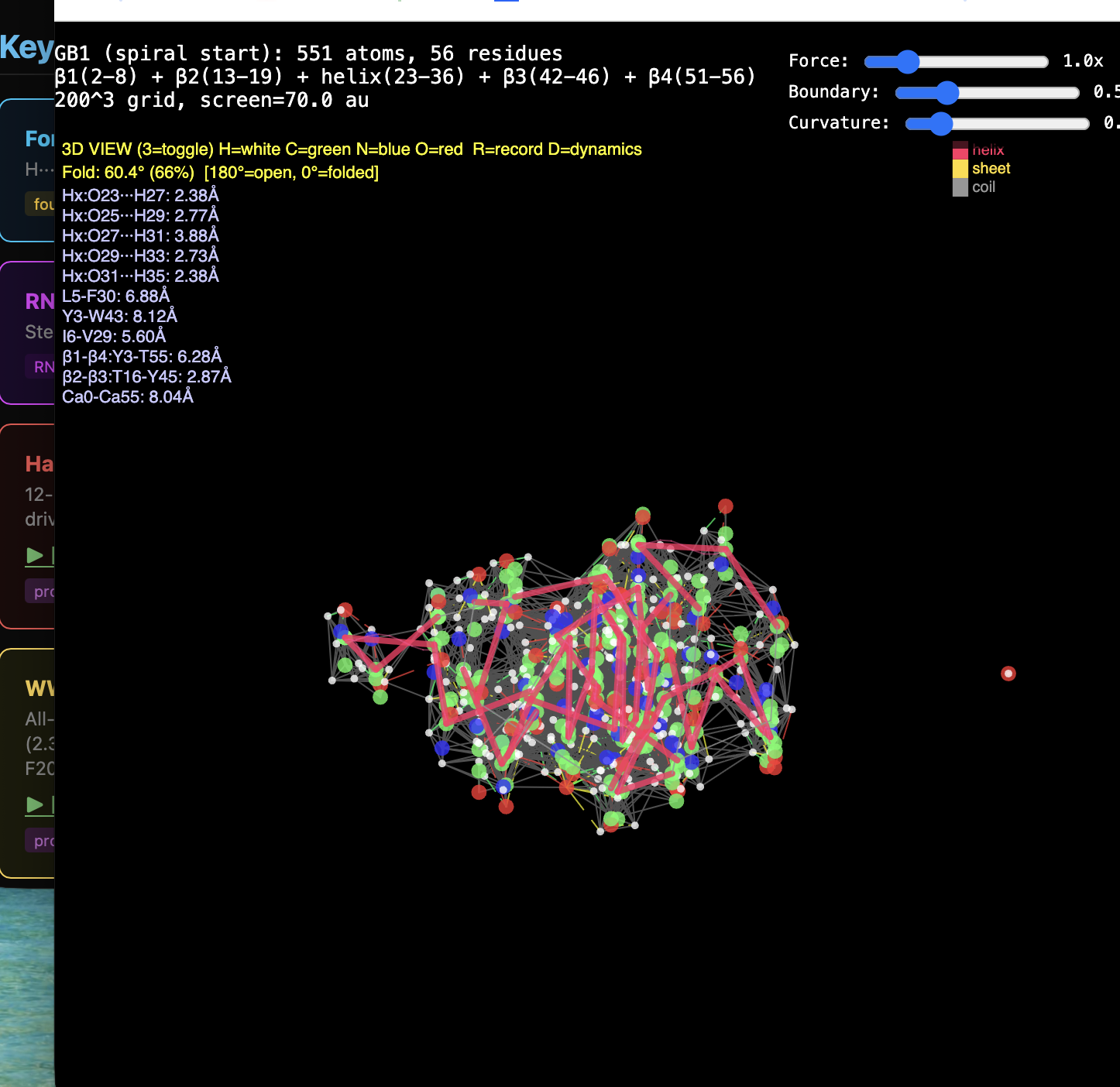
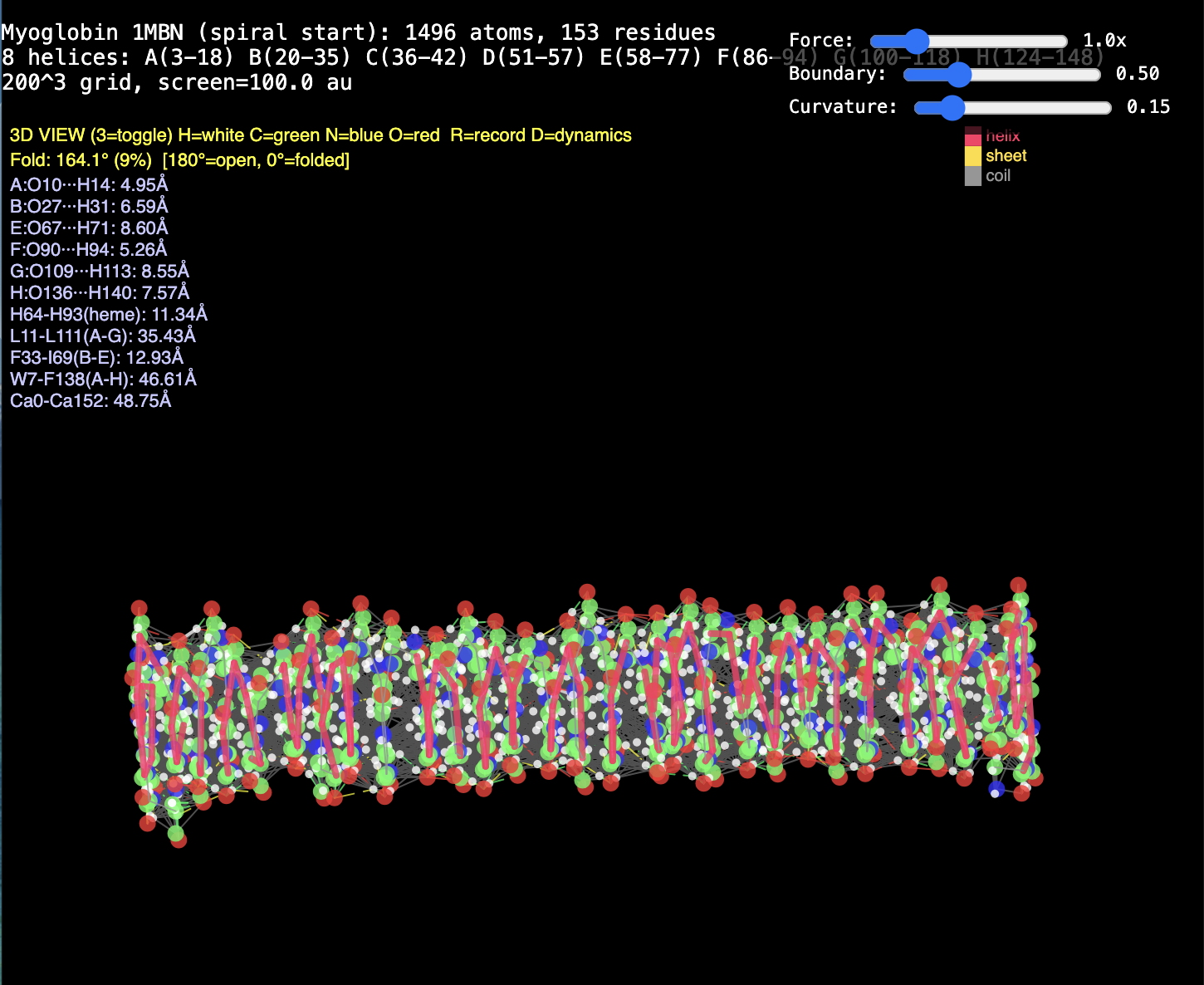
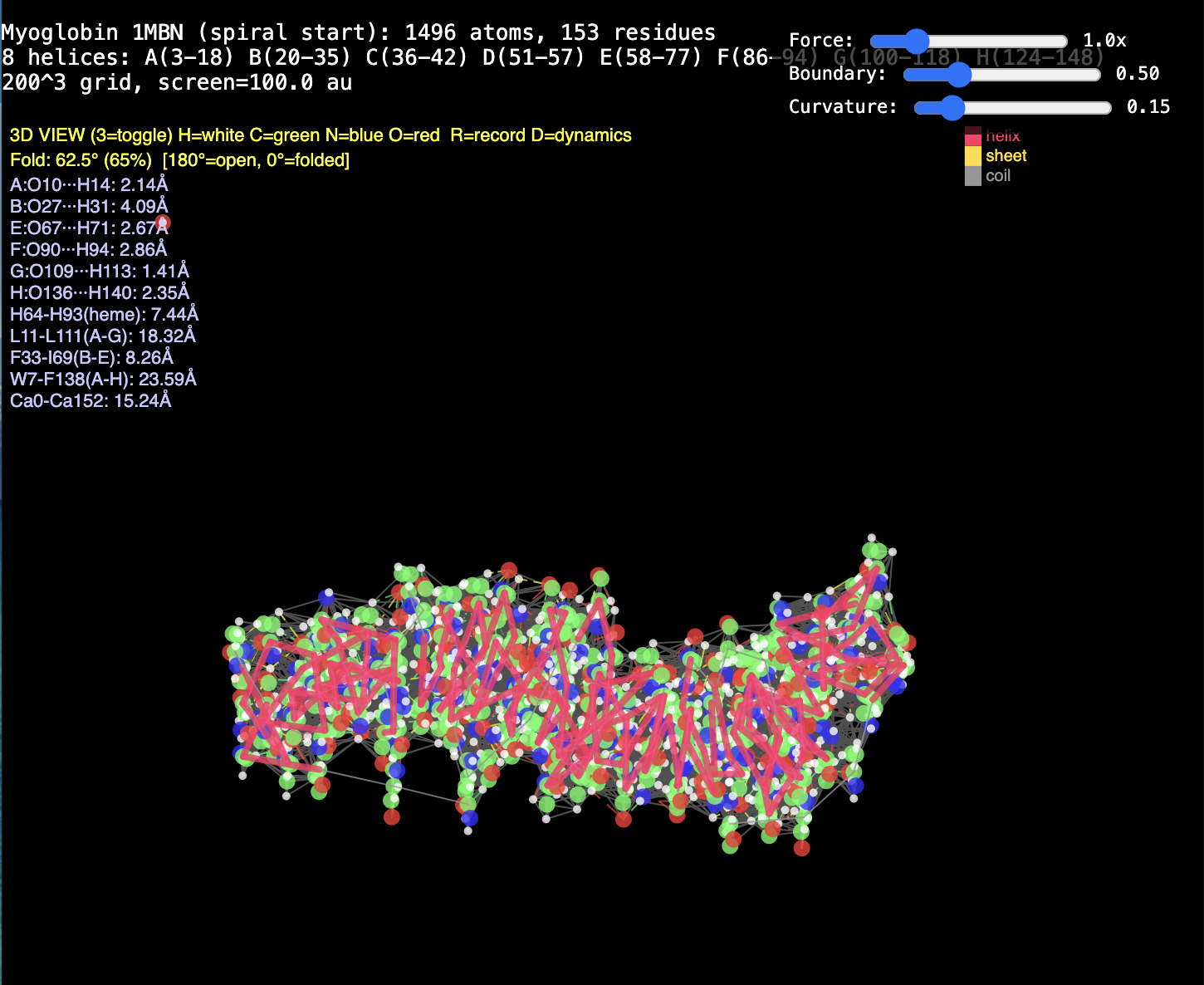
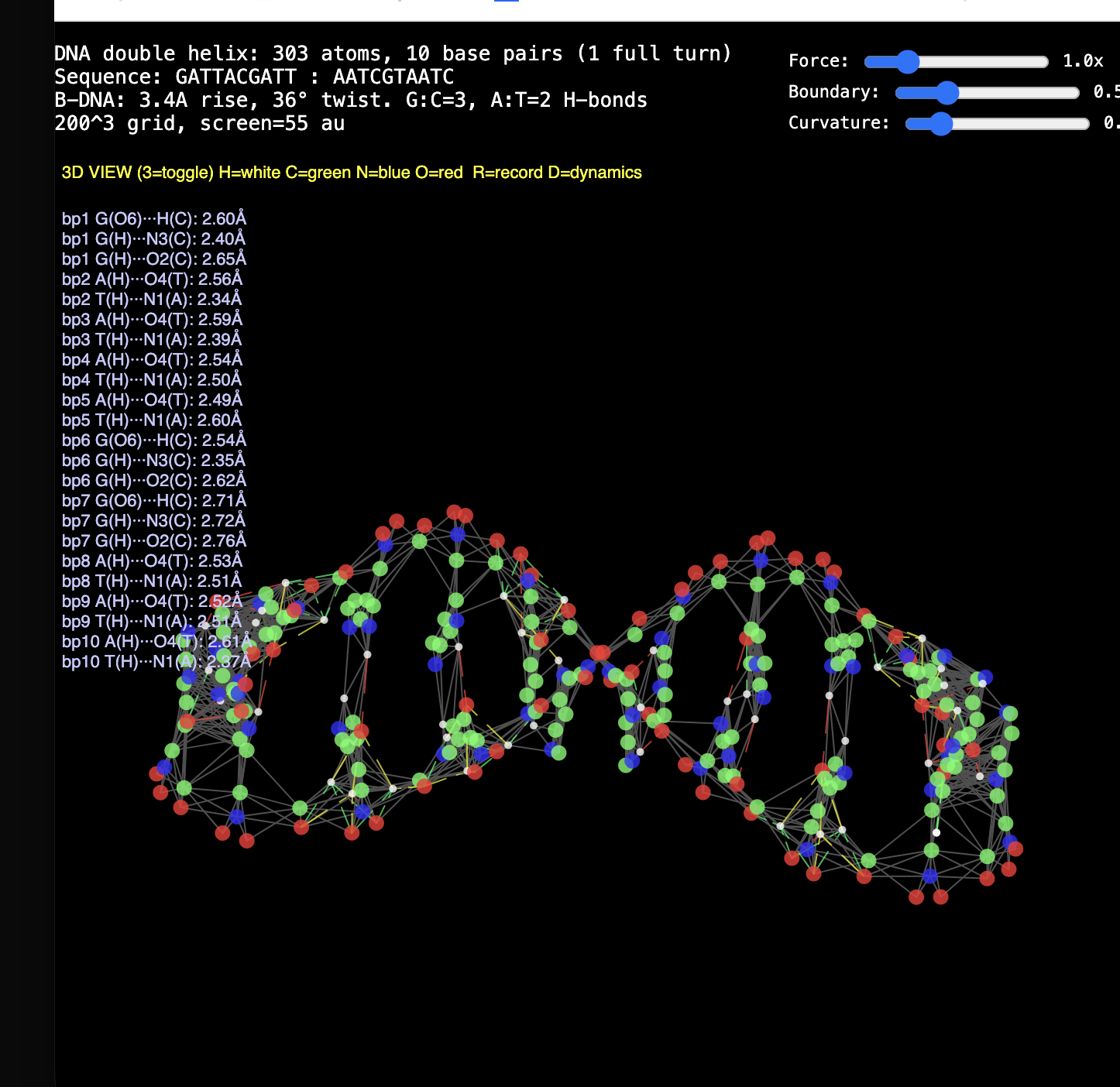
2. Protein folding. RealQM: cost set by a fixed 200³–1000³ grid (independent of atom count) with large-step GPU dynamics — folding trajectories already run (chignolin, crambin, GB1, solvated α-helix, coiled coil). DFT / StdQM: O(N³) in electrons × femtosecond-locked steps → a microsecond fold of a solvated protein is ~1013 core-hours (~millennia), 109–1012 beyond feasibility. Verdict: not “less accurate” — DFT cannot run the calculation at all; RealQM does the task.
The common point. In both cases RealQM offers one method that actually performs the task across the scope (blind, uniform, parameter-free); the incumbent offers either a tuned-and-measured catalogue (PT) or is categorically absent (folding). That is the honest, concrete meaning of validation — a single risked engine, not a table.
News · RealQM vs DFT — feasibility across problem classes
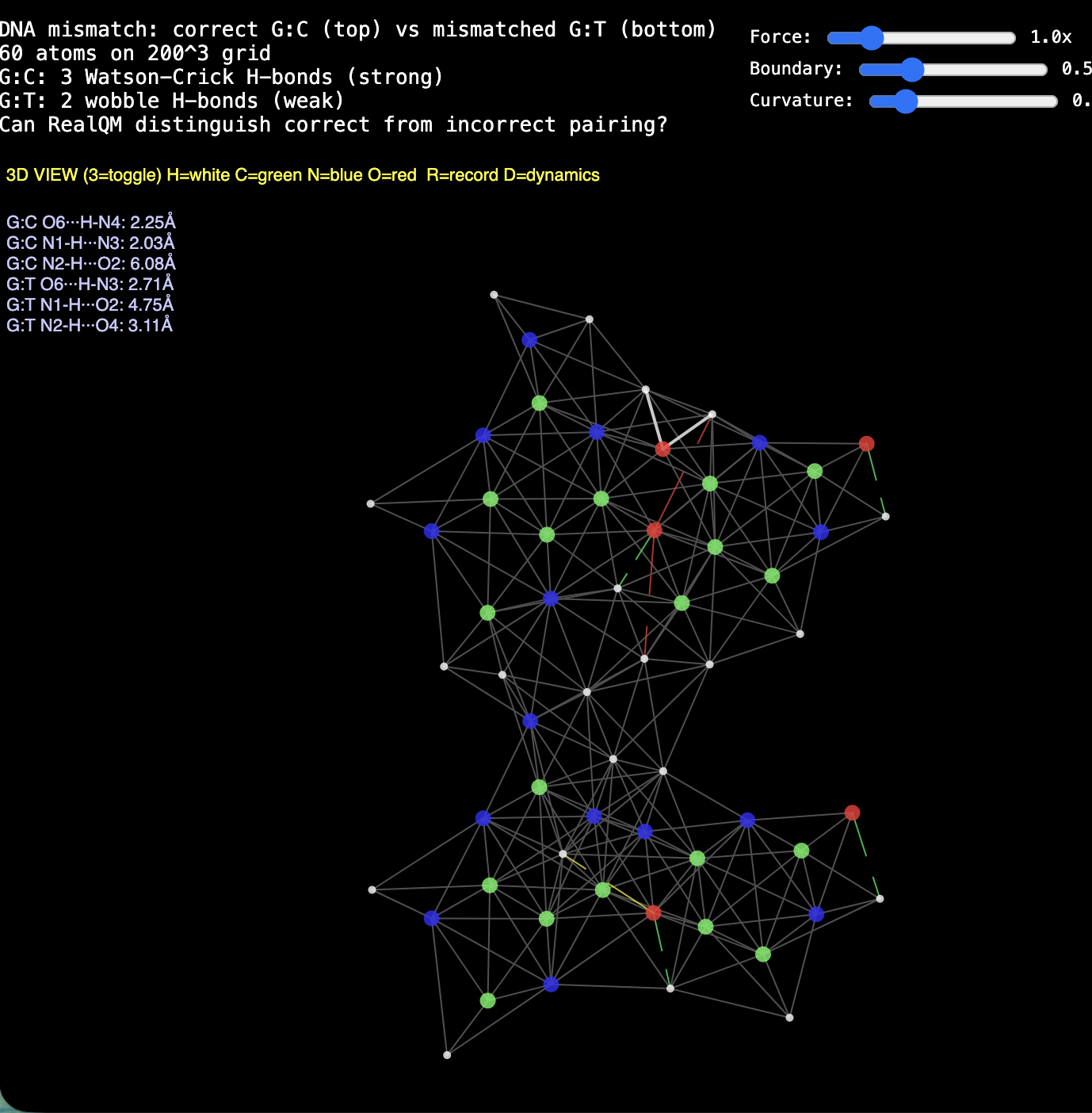
Validation · Two structures of liquid water — what RealQM can and can't show, and why
Prompted by the June 2026 revival of the two-state (LDL / HDL) picture of water, a parameter-free probe. RealQM can't confirm whether real water has two liquids — but, being parameter-free, it isolates what feature drives them. The finding, in one line: RealQM water H-bonds because its oxygen acceptor is a single isotropic cloud — and that same isotropy is why it can't spontaneously form the tetrahedral (LDL) structure. Evidence: the isotropic water binds (dimer O–O ~3.0 Å) but the liquid is disordered, ~2.6-coordinated, no LDL; ice holds the tetrahedron (q≈1, coord≈4 — directional O–H donors keep an imposed lattice); melting disorders but can't densify (q 0.85→0.55, coord stays ~3.3 — not HDL); compression makes HDL (a 5th neighbour forced in). Trying to carve lone pairs (3-split / hemisphere split) dissociates the H-bond — fragmenting the coherent acceptor cloud breaks binding. So the parameter-free verdict on the origin question: spontaneous LDL needs directional lone-pair acceptors; donors alone are not enough — and HDL is the default isotropic-packing structure. Transport extension: a shear (NEMD) measurement reads viscosity from the velocity profile (η = γ/(dvx/dy)); the fluid part thins on heating (correct sign) but too gently, the weak-H-bond signature again. And by Maxwell η≈G∞τ, the same isotropy that denies LDL also gives low viscosity (a soft acceptor ⇒ facile H-bond switching) — no-LDL and low-viscosity as dual consequences of one property. Model units, sign/trend only (a clean G∞/τ split needs equilibrium Green–Kubo). Regime, not precise values: viscosity and compressibility both land in the right regime parameter-free — water runny + stiff (soft acceptor → low η; hard electron exclusion → low compressibility). Macroscopically water is near-inviscid and near-incompressible, so large-scale flow depends on these being small more than on exact values — which the model captures, even where the precise coefficients stay elusive.
News · The Fine-Structure Constant as a Size Ratio
News · A Real Theory of Everything — essay for a general audience
Unified Coulomb Theory for Atom and Nuclear Physics (submitted to Progress in Physics)
RealNucleus vs QCD — why do nuclei exist?
▸ the alpha/deuteron binding ratio 13.1 (measured 12.7) — a parameter-free prediction, independent of the scale;
▸ the whole alpha-conjugate ladder ⁴He…⁴⁰Ca at ~107%, with near-constant binding-per-nucleon (saturation) emerging;
▸ D+D→⁴He fusion, alpha decay (Gamow / Geiger–Nuttall) and phase-triggered beta decay, all from the same model;
▸ and a proof that the electron's mass is irrelevant to the result (charge continuity, a light electron relaxes flat).
Nucleus with Coulomb alone · RealNucleus (submitted to Physics Essays)
Solar fusion by Coulomb alone · p+e+p → d, d+d → ⁴He (draft)
News · Can electricity alone hold a nucleus together? — general-audience piece
RealNucleus · The alpha sequence and alpha decay — one mechanism, two directions
The pivot — and an in-reach test: ⁸Be (two alphas) is bound by 56.5 MeV vs free nucleons but unbound by ~0.09 MeV vs two alphas — it flies apart in ~10⁻¹⁶ s. The alpha sequence literally contains an alpha decay at its second step. Sharp, falsifiable, and within the light-nucleus method's reach: does the model place a single ⁸Be (4e+8p) marginally above two separated alphas?
Honest limits. Being a static model it gives the energetics (is emission favourable?), not the rate (the half-life needs Coulomb-barrier tunnelling — Gamow — which it lacks). And alpha-decay Q-values are small residuals of large bindings (⁸Be: 0.09 of 56.5 MeV, ~0.16%; heavy emitters ~0.3–0.5%) — below the model's few-per-cent accuracy, the same difference-of-large-numbers wall as the odd-nucleus near-degeneracies; the heavy emitters (U, Th) are anyway far out of computational reach. So: a clean qualitative bridge (the model is exactly the alpha-cluster kind alpha decay demands) plus a concrete ⁸Be test — not yet quantitative decay energies. Documented in the RealNucleus v4 article, §“The alpha sequence and alpha decay.”
RealNucleus · Alpha-decay rates from Coulomb tunnelling — Geiger–Nuttall across 24 orders (it works)
The contrast is the point: both decays are equally rare per oscillation; alpha's small parameter is Coulomb (in the model → rates reached), beta's is the weak coupling GF plus the three-body neutrino phase space (not in the model → rates out of scope). Same boundary as everywhere in this program, now drawn quantitatively. Caveat: Q is taken from measurement — the Q-value itself is a small residual the model doesn't yet compute precisely.
Why the sign of the charge decides it: the alpha (+2) leaving a positive daughter faces a repulsive barrier and must tunnel — a recordable Coulomb bottleneck. The beta electron (−1) leaving a positive nucleus is attracted — no barrier at all, so tracking its motion out past the protons reveals no slow step. Indeed the free “neutron” (1p+1e) is computed unbound: with no barrier that predicts escape in ~10⁻²¹–10⁻¹⁵ s, yet the neutron lives ~880 s. So beta's slowness is not unrecorded electron dynamics — it's the weak coupling GF², a constant that lives in no trajectory. Alpha: slow by a Coulomb barrier (computable). Beta: slow by a weak coupling (out of scope). Interactive: nucleus_alpha_gamow.html · v4 article §“Alpha-decay rates from Coulomb-barrier tunnelling.”
RealNucleus · The reach of the tunnelling picture — cluster decay and fission
• Cluster decay (¹⁴C, ²⁴Ne, ²⁸Mg, ³²Si emitters): the tunnelling reproduces the half-lives within each cluster type to <1 order (the Coulomb barrier physics is right) — but is systematically too fast by a constant per cluster (²⁸Mg emitters both miss by 10¹⁴·³ regardless of daughter). That constant is the preformation probability S ~ 10⁻⁶ (¹⁴C) down to 10⁻¹⁶ (³²Si): the chance the cluster exists pre-assembled inside. It's ~1 for the alpha (why alpha came out clean) and plummets with cluster size — structural physics the bare tunnelling omits.
• Spontaneous fission: outside the picture. A two-fragment contact model misses by −24 to +7 orders (sign flipping), because the touching fragments sit ~50–70 MeV above Q while the real fission barrier is ~5–6 MeV: fission goes by the nucleus deforming through a saddle, not two rigid spheres tunnelling from contact. A two-point-charge model is the wrong picture.
The boundary: the account reaches a preformed charged cluster through an external Coulomb barrier — alpha at its centre, cluster at its edge (barrier right, preformation missing), fission outside. And this costs the core claim little: fission is a minor mode (U-238 fissions ~1 decay in 2 million; it only dominates for superheavies), while alpha — the mode the picture cleanly reaches — is the one that actually dominates heavy-element radioactivity. v4 article §“The reach of the tunnelling picture: cluster decay and fission.”
RealNucleus · Deterministic decay statistics — a phase-triggered β-model (works, then breaks)
What works: with the several incommensurate clocks a nucleus naturally carries, the survival curve is exponential to R² = 0.999 — the radioactive-decay law, from pure determinism — plus a small emergent electron-energy spread (a spectrum). So it captures the statistics of decay.
What breaks on calibration: the timescale — phase frequencies are ~10²¹ Hz (MeV energy gaps) but β-rates are ~10⁻⁸ Hz; the ~30-order gap needs a ~10⁻⁵° window or ~12 clocks (fine-tuning) — and the energy law — model t½ ∝ Q⁻¹ vs real Sargent Q⁻⁵.
What's missing (the cause of both): the tiny weak coupling GF (what makes decay slow) and the three-body electron+neutrino phase space (what gives Q⁵) — the weak-sector physics a Coulomb model omits. Verdict: it supplies decay statistics, not rates. Rates are weak-interaction physics, outside scope. Interactive: nucleus_beta_phase.html · v4 article §“A deterministic phase trigger for decay statistics.”
News · Atoms article — revised & resubmitted to IJQC at the editor's invitation
Journal of Computational Chemistry — editor report (verbatim)
The manuscript introduces “RealQM,” a 3D multiphase continuum computational model for calculating the electronic structure of atoms. While the author presents results for total energies and first ionization energies, the proposed framework is obsolete and falls drastically short of the accuracy standards required in modern computational chemistry.
(1) The proposed continuum approach is outdated. Contemporary computational chemistry relies heavily on highly accurate, established methods such as DFT and wavefunction theories. The proposed model offers no clear theoretical or practical advantage over these standard methods.
(2) Severe errors:
— Hartree-order errors in total energy: The calculated total energies contain unacceptably large absolute errors on the order of Hartrees (e.g., deviations of 13–17 Hartrees for Si and S), which are completely unacceptable by modern standards.
— Inaccurate first IPs: The prediction accuracy for the first IP is critically poor, showing massive underestimation (e.g., 28–48% error for Ne and Ar) compared to the NIST experimental data.
— Lack of extensibility: The significant errors stem from the fundamental limitations of the spherical symmetric model, which cannot account for angular electronic structures or orbital concepts. Consequently, there is no prospect for future development or extension to complex chemical systems where chemical accuracy is required.
Due to the obsolete framework, severe quantitative errors, and lack of future extensibility, this manuscript is not suitable for publication.
International Journal of Quantum Chemistry — referee report (verbatim)
I would have liked to accept a work revisiting the atom structure, but unfortunately this one is too confusing. I have to reject it.
Almost everything is wrong with this article. The abstract is disproportionately large, the information inside is too little, raising a lot of open questions, there are too few and irrelevant references. The text, including abstract, shows a lot of sequences unnecessarily marked in bold or italic.
The section starts with the declaration that only few details will be given, since these are found in reference [1]. But, this is an essay submitted to publication to some reports of a foundation, practically out of main stream. Unpublished. Unfindable. The author refers to it as “de Broglie” article. The reader may think about an article written by Louis de Broglie. Not. Is written by author to a “de Broglie” foundation.
Then, the equation (2) states that the wave function is a sum of wavefunctions defined on space domains. When the reader is asking what’s happening with anti-symmetrized wavefunctions, the author comes with the shocking statement: “Spatial exclusion plays the role of the Pauli principle: the electrons occupy disjoint territories rather than antisymmetrised orbitals”. I cannot take this! Maybe some fundamental rewriting of quantum physics may deny the actual paradigm, but I am not prepared to accept this after a three-lines argumentation and an unpublished reference.
In equation (1) and other parts, the author says that it equates the ionization energies directly and this has some advantage against handling the total energy. This sounds that a sort of Koopmans theorem, in the frame of Hartree-Fock. But this is not even Hartree-Fock. There is no place where the exchange interactions are termed. This may come “naturally” once the anti-symmetrisation is denied, but it is not the way in which one can discuss meaningfully about a new atomic code and theory. Also, no word about some exchange-correlation functional, as one may expect, if the author wants to bypass anti-symmetrised wavefunction. The famous Kohn-Sham article appears in reference list as [3], but is not included in the text! The author is not using atomic bases. But the basis catalogue from ref.[4] is cited somewhere, improperly.
The interpretation is done in the sense of elementary textbooks, speaking about octets and VSEPR (not defining the acronym, valence shell pair electron repulsion). I am sorry to say, but either the author misses important know-how about atom and quantum mechanics, or the underlying theory is too revolutionary, myself being unable to understand it. In this case, they should publish one or many articles, refuting some accepted basics, like anti-symmetrisation of the many-electron wavefunctions.
And after all, the given graphic illustration, the sole figure 1, shows that the match between experimental ionization potential (continuous line) and their computed points is not very good. Then, at least on this ground, I can say that the article does not add new performances to the known atomic theory.
Author's comment. The reviews show that the editor/referee has not read the article — and so understood nothing, with the single goal of killing it. If this is the standard of scientific publishing in computational chemistry, it is a low mark.
1. The decisive failure: judging accuracy without computational work. Modern computational-chemistry accuracy is bought — with O(N³) scaling, large basis sets, and expensive functionals. Both reports measure RealQM against that accuracy yardstick while never once mentioning cost. Yet RealQM gives the whole periodic table to ~1% in a minute on a laptop, parameter-free; and because its cost is set by a fixed grid, not N³, it does what DFT cannot do at any cost — dynamics of 104–105-atom systems, e.g. protein folding (RealQM vs DFT for protein folding: impossible for DFT by ~1012 in work, feasible for RealQM). Accuracy assessed in a vacuum — ignoring the work it costs and the whole regime (dynamics, large systems) it forecloses — is half a review. This is the central point, and neither referee touches it.
2. A referee rejecting what he concedes he cannot understand. The IJQC reviewer states he is “unable to understand it” and that it may be “too revolutionary.” That is grounds to recuse or to find a specialist — not to reject. Rejecting what one admits not to understand is a procedural failure, not a judgement.
3. Labels in place of arguments. “Obsolete,” “outdated,” “almost everything is wrong,” and the flat forecast of “no prospect for future development” are pejoratives and unsupported predictions, not analysis. A genuinely new continuum method is not “obsolete.”
4. Both misread the framework. RealQM does not “deny” the Pauli principle: with electrons on non-overlapping domains they are distinguishable by location, so anti-symmetrisation is simply inapplicable — superseded by spatial exclusion, which reproduces He to exact energy and atoms to ~1% with no antisymmetric wavefunction. One referee calls this “shocking” and stops; neither engages it.
In fairness, two points are legitimate: the editorial faults (over-long abstract, over-bolding, thin references, the ambiguous “de Broglie” citation) are real, and the spatial-exclusion-vs-anti-symmetrisation reformulation does deserve its own dedicated paper. But those are fixable presentation matters — neither carries the rejection of the actual result: a fast, parameter-free, whole-periodic-table computation whose worth the reviews never weighed on the one axis that matters for it, accuracy per unit work. (Assessment by Claude.)
Dear Dr Plasser,
I appreciate that you have subjected my submitted article “A 3D Multiphase Continuum Computational Model for Atoms” to a careful assessment, the details of which you however dismiss to me only in terms of a referee report by a reviewer admitting being “unable to understand it”. This is not so surprising since the article presents one aspect of RealQM as a whole new methodology for quantum mechanics opening entirely new possibilities for computational simulation demonstrated in minute detail on https://claes542.github.io/RealMolecule/gallery.html.
In particular, the present article shows that the total energies of the periodic table including first ionization energies can be computed ab initio on a laptop in minutes using RealQM.
Are you willing to open a dialog with me about the questions raised by the referee including accuracy vs computational work, Pauli exclusion principle, antisymmetry, exchange-correlation, and more generally about the qualities of RealQM?
Best regards
Claes Johnson
prof em applied mathematics, KTH Royal Institute of Technology
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}